Inferring Model Parameters in Systems Biology
Samuel Bernard bernard@math.univ-lyon1.fr
September 4th, 2019
Inferring Model Parameters in System Biology
Dataset with \(n\) observations \(y_i\) at time points \(t_i\), \(i=1,...,n\), and a model function \(m(t; \theta)\) that depends on time and on a certain number of parameters \(\theta = (\theta_1, \theta_2, \theta_k)\). Can we find values for \(\theta\) such that the function \(m\) will take values close to \(y_i\) when evaluated at \(t_i\):
\[
m(t_i; \theta) \sim y_i
\]
How to choose the model
If the number of parameter \(k\) equal to or larger than \(n\), it is in general possible to match exactly all the data: \(m_i = y_i\). This is called interpolation.
When the number of parameters if smaller than \(n\), it will be in general not possible to match all data points, and there will be a trade-off to achieve between being far-off for few data points and close to the others, or being a bit further away from all data point.
The way to resolve this trade-off is to pose a minimization problem. That is, we will define a scalar-valued objective-function \(f\) that depends on \(m\) and \(y\) and has the property that \(f(y,y) = 0\). By far the most common objective-function used in biology is the sum of squares of the error between \(m_i\) and \(y_i\): \[
f(m,y) = \sum_{i=1}^n (m_i - y_i)^2 = ||m - y||^2
\]
Small is beautiful
We randomly generate n=15 pairs of points, \(x\) in [0,1] and \(y\) in [-1,1].
n <- 15
x <- runif(n) # generate n random numbers in [0,1]
y <- runif(n, min = -1, 1) # generate response in [-1,1]
plot(x,y)
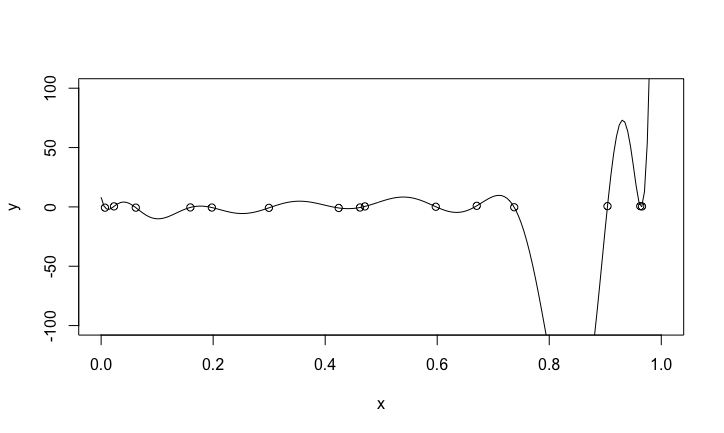
Polynomial interpolation
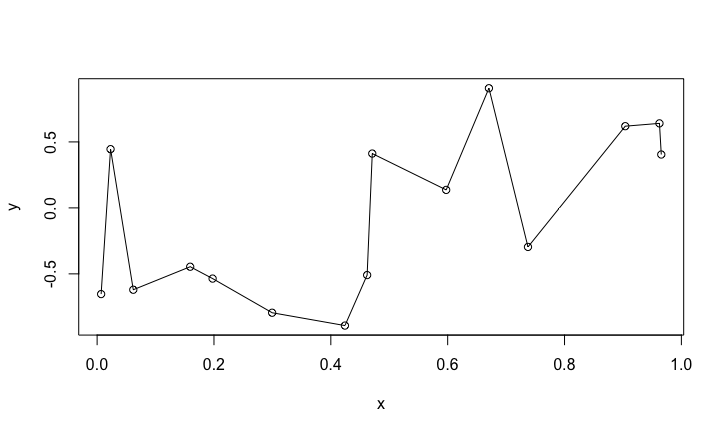
Now we fit a polynomial of degree 14 (it has 15 coefficients/parameters)
interp <- lm(y ~ poly(x,n-1))
plot(x,y)
sx <- sort(x)
lines(sx, predict(interp, data.frame(x = sx)))
The fit is perfect, the polynomial function goes through all the data points. Suppose we get a new pair of data \((x,y)\). How good will be the prediction?
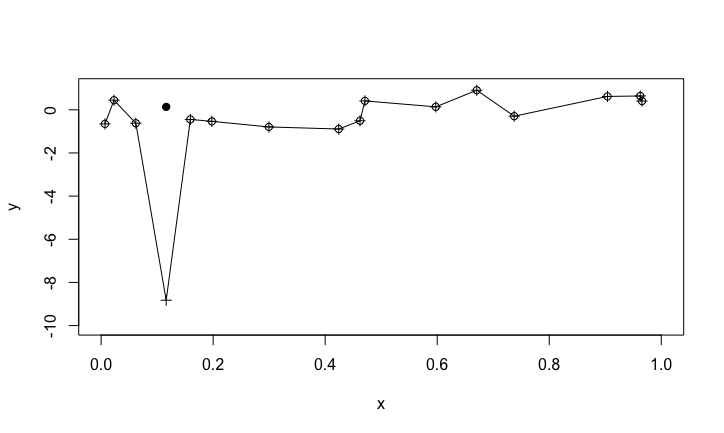
Prediction
newx <- runif(1)
newy <- runif(1) # add a new data point
plot(c(0,1),c(-1,1), type = 'n', xlab = 'x', ylab = 'y')
points(x,y)
points(newx,newy, pch = 19)
lines(sort(c(x,newx)), predict(interp, data.frame(x = sort(c(x,newx)))))
points(sort(c(x,newx)), predict(interp, data.frame(x = sort(c(x,newx)))), pch = 3)
What the polynomial really looks like
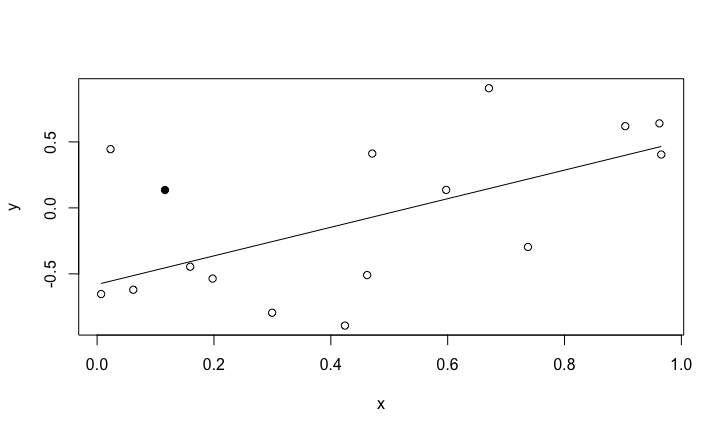
Linear regression
linreg <- lm(y ~ x) # linear regression
plot(x,y)
points(newx,newy, pch = 19)
sx <- sort(x)
lines(sx, predict(linreg, data.frame(x = sx)))
The linear regression has two parameters, (intercept and slope). Between 2 and 15 parameters, there is probably a optimal number of parameters that can be fitted for a given dataset.
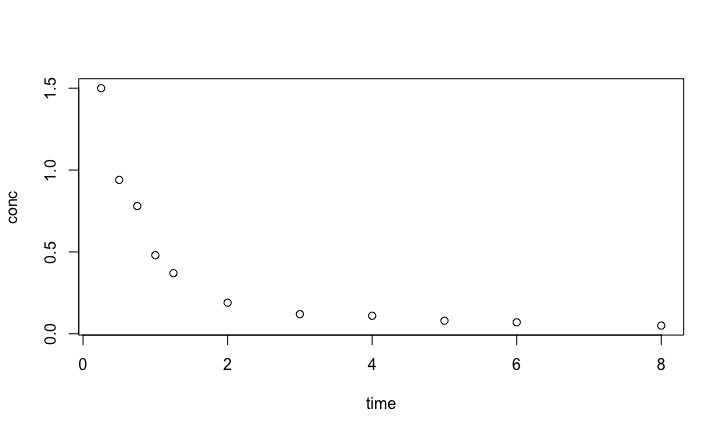
Pharmacokinetics
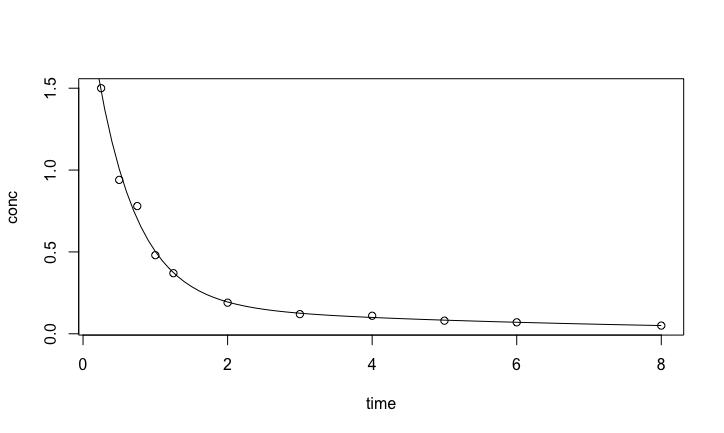
We use the pharmacokinetics of Indomethacin dataset Indometh from the datasets package in R.
Indo.1 <- Indometh[Indometh$Subject == 1, ]
with(Indo.1, plot(time,conc))
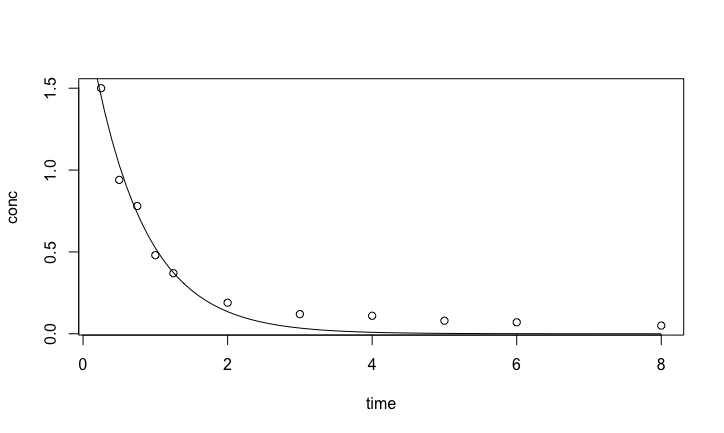
Nonlinear fit with a known model function
We perform a nonlinear least-square fit with the routine nls from R. The response variable is the concentration conc, the independent variable is time, and the model is \[
m(t) = c_0 \exp( - k t)
\] with two parameters \(c_0\) and \(k\).
indometh.nls <- nls(conc ~ c0 * exp(- k * time), # model
data = Indo.1, # dataset
start = list(c0 = 1, k = 0)) # initial guess for the parameters
lines(seq(0,8, by = 0.1),predict(indometh.nls, list(time = seq(0,8, by = 0.1))))
Result of the nonlinear model
Formula: conc ~ c0 * exp(-k * time)
Parameters:
Estimate Std. Error t value Pr(>|t|)
c0 2.0332 0.1392 14.60 1.42e-07 ***
k 1.3563 0.1232 11.01 1.60e-06 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.07371 on 9 degrees of freedom
Number of iterations to convergence: 10
Achieved convergence tolerance: 8.285e-06
Result of the nonlinear model
Nonlinear regression with an ODE model
To have a basis for comparison, we use the same dataset and the same model, except we express the model as an ODE
\[
\frac{dC}{dt} = - k C
\] with initial condition \(C(0) = c_0\). The code is a bit more complex but the results is identical to the exponential model
Formula: conc ~ odemodel(time, c0, k)
Parameters:
Estimate Std. Error t value Pr(>|t|)
c0 2.0332 0.1392 14.60 1.42e-07 ***
k 1.3563 0.1232 11.01 1.60e-06 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.07371 on 9 degrees of freedom
Number of iterations to convergence: 7
Achieved convergence tolerance: 3.348e-06
Refining and selecting a model
The fit with the exponential model is quite good at initial times, but after 2 hours, the concentration decreases more slowly than the fitted exponential. This may be because there is residual amount of drugs in peripheral tissues with slower elimination rates. A bi-exponential can take care of the two section of the pharmacokinetic data \[
b(t) = c_{01} \exp ( - k_1 t ) + c_{02} \exp( - k_2 t )
\]
Fitting the bi-exponential model
The R code is
biexpmodel <- function(Time,c01,c02,k1,k2) {
return(c01*exp(- k1 * Time) + c02*exp(- k2 * Time))
}
with(Indo.1, plot(time,conc))
indometh.biexp <- nls(conc ~ biexpmodel(time,c01,c02,k1,k2), # model
data = Indo.1, # dataset
start = list(c01 = 2, c02 = 1, k1 = 2, k2 = 1)) # initial guess for the parameters
lines(seq(0,8, by = 0.1),predict(indometh.biexp, list(time = seq(0,8, by = 0.1))))
Result with the bi-exponential model
The fit looks much better! That the fit is better is to be expected, the bi-exponential includes as a subset the exponential model. So, it is really better? One way to test this is with the Akaike Information Criterion.
Exercise 1
The bi-exponential model can also be expressed as the solution of a system of ODEs. How could it look likes?
Exercise 2
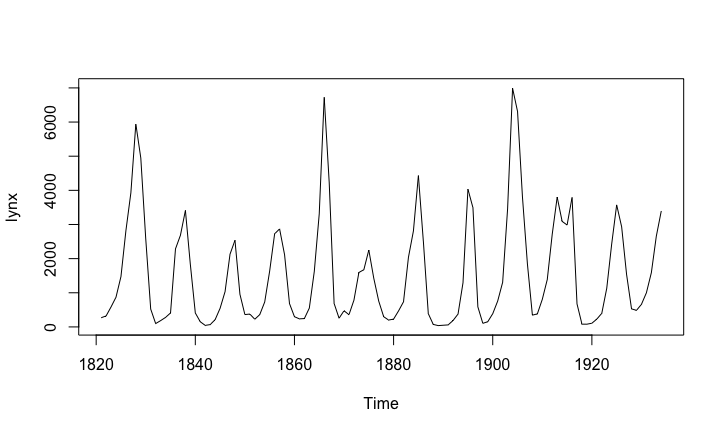
The figure shows the number of lynx in trapped in Canada between 1821 and 1934. If you were a planner, trying to manage the lynx population in a sensible way, how would you do?
Exercise 3
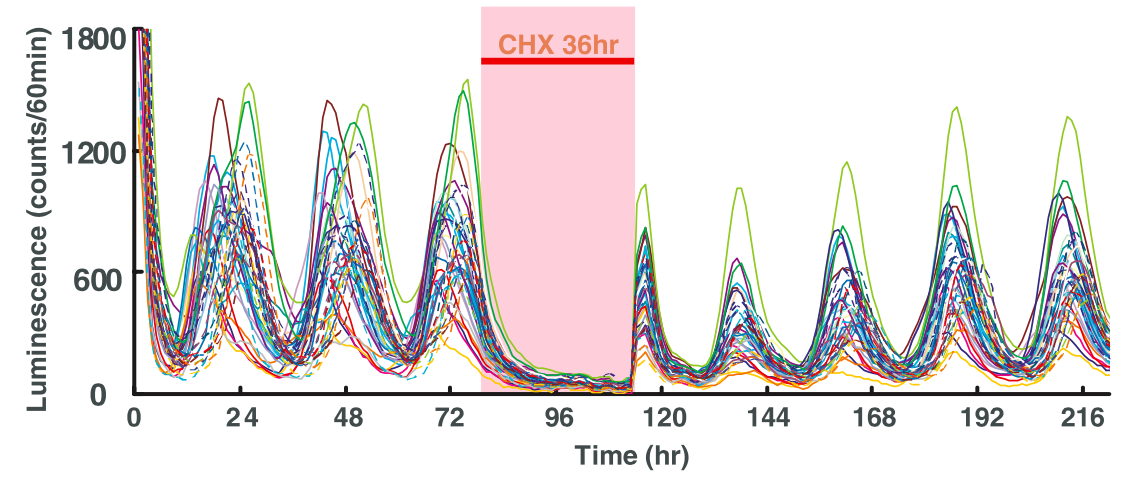
The figure shows the bioluminescence of a luciferase reporter protein with a mPer2 promoter. How would you design a mPer2 model?
References
MJ Crawley, The R Book, 2012, Wiley